

To download a copy of this form, click here
Participant Information Sheet
A Double-Blind, RandoMised, Placebo-Controlled Study to Assess the Efficacy and Safety of oRal deliVery of sodium Pentosan Polysulfate (PPS) compared to placebo in participants with symptomatic kneE osteoarthritis (OA) and dysLipidemia
Title
Short Title
MaRVeL Study
The University of Sydney
Project Sponsor
Professor David Hunter
Osteoarthritis Clinical Research Group
Affiliation: Kolling Medical Research, The University of Sydney
Principal Investigator
Invitation
You are invited to participate in a research study to investigate the effect of an oral medication, Pentosan Polysulfate (PPS) compared to placebo on pain in participants with symptomatic knee OA and dyslipidemia (the imbalance of lipids such as cholesterol, low-density lipoprotein cholesterol, (LDL-C), triglycerides, and high-density lipoprotein (HDL).
The study is being conducted by Professor David Hunter, Florance and Cope Chair of Rheumatology, Professor of Medicine, The University of Sydney, at Royal North Shore Hospital (RNSH), St. Leonards.
Before you decide whether or not you wish to participate in this study, it is important for you to understand why the research is being done and what it will involve. Please take the time to read the following information carefully and discuss it with others if you wish.
Participation in this research study is voluntary.
By giving your consent to take part in this study you are telling us that you:
• Understand what you have read.
• Agree to take part in the research study as outlined below.
• Agree to the use of your personal information as described.
If you agree to participate in this study, you will be asked to sign the Participant Consent Form electronically. You will be provided a copy of the Participant Information and Consent Form via email to keep for your records.
Location
Department of Rheumatology, Clinical Administration 7C,
Royal North Shore Hospital, St Leonards, NSW, 2065
PART 1 What does my participation involve?
1. ‘What is the purpose of this study?’
entosan Polysulfate (PPS), the active ingredient, is derived from the beech wood hemicellulose (the bark of the Beech tree). It has been approved by the Therapeutic Goods Administration (TGA) in Australia and the Food and Drug Administration (FDA) in the US (commercial name - ELMIRON®) for more than twenty years as an oral medicine for interstitial cystitis (chronic inflammation of the bladder wall). It has been used in Europe for many years as a mild blood thinner. It has also been reported to have a lipid-lowering property. This drug is NOT approved by TGA for osteoarthritis, and we are conducting this study to understand the potential role of Pentosan Polysulphate in providing pain relief for people with knee osteoarthritis.
Currently, treatment of osteoarthritis is primarily aimed at easing symptoms. However, it is becoming more important to identify factors that may be related to the worsening of symptoms, such as poor blood supply to the knee joint and cartilage, which may be responsive to treatment. This study aims to establish whether Pentosan Polysulfate can improve the knee pain, decrease the cholesterol levels in the bloodstream and improve blood circulation to the knee joint. It is a double-blind, placebo-controlled, phase II trial where participants will be randomised to receive the active PPS or placebo capsules based on group allocation.
“Randomised trial”: Sometimes doctors don’t know the best way of treating patients with a condition, so comparisons need to be made between different treatments. To do this, study participants are placed into groups and given different treatments; then, the results are compared to see if one treatment is better than the other. To ensure the groups are alike to start with, a computer allocates each study participant into their group randomly, like the flip of a coin. Neither the study staff nor the study participant can decide which treatment the participant receives.
“Blind trial”: In a “blind trial” the study participants do not know which treatment group they are in. If the trial is “double-blind”, neither the researcher nor the study participant knows which treatment the participant is receiving (although, if the researcher needs to find out, they can do so).
“Placebo”: A placebo is a dummy treatment that looks like genuine medicine but contains no active ingredient.
The University of Sydney conducted a pilot, single arm, open label study in 2019-2020 and information from this pilot study has served as a basis for this randomized trial that aims to improve long-term outcomes such as pain and stiffness in people with knee osteoarthritis.
2. ‘Why have I been invited to participate in this study?’
You are being invited to participate in this study because you are diagnosed with knee OA and/or have experienced symptoms associated with OA for at least 6 months and your doctor told you that you have an elevated blood lipids level.
Knowing what is involved will help you decide if you want to take part in the research. Please read this sheet carefully and ask questions about anything that you don’t understand or want to know more about. This Participant Information Statement tells you about the research study.
3. What if I don’t want to take part in this study or if I want to withdraw later?’
It is completely up to you whether or not you participate. If you decide not to participate, it will not affect the treatment you receive now or in the future. Whatever your decision, it will not change your relationship with the staff caring for you.
New information about the treatment being studied may become available during the study. You will be kept informed of any significant new findings that may affect your willingness to continue in the study.
If you wish to withdraw from the study once it has started, you can do so at any time without having to give a reason.
4. ‘What does this study involve?’
The MaRVeL Study is a 6-month single site, randomised, placebo-controlled, double blinded trial conducted in Sydney. You will be required to attend five (5) visits to Royal North Shore Hospital and two (2) visits for MRI scan to Cremorne, NSW.
You must be willing to attend the following visits for the study:

* All participating Northern Sydney NSW Health pathology collection centres locations will be provided
You must follow all instructions given to you while you are participating in this research project. If you are unsure about what you are supposed to do, ask the study research coordinator.
You must not take part in any other study while you are taking part in this research project.
PART 2 Study eligibility and treatment
1. Screening Period
The screening period will last for up to six weeks. The purpose of it is to make sure that the study is right for you.
Online screening
If you are interested in this trial, you will be required to complete an online screening survey to assess your eligibility. You may be contacted by phone or email if further information is required.
Phone Screening
If you are deemed potentially suitable after completing the online screening, the study coordinator will go through a second screening over the phone.
• The study coordinator will check/confirm the details of your medical history, your OA symptoms and will explain the research project to you and answer any questions you may have.
• You must tell us details of medications that you have been taking (within 30 days before the screening visit). It includes prescription medicines, vitamins, minerals, and medications that do not require a doctor’s prescription.
Prohibited Medication
Some medications are not allowed.
• You must be willing to stop using NSAIDs (anti-inflammatory drugs, e.g. Nurofen, Celebrex, Mobic, etc.), high doses of aspirin (>325mg), opioids (such as Tramadol) and steroids (Voltaren), except creams or gels applied to the skin. You cannot treat pain in your index knee with Voltaren while participating in the study.
• You cannot take part in the study if you are taking blood thinners.
It is important that you read and understand this information carefully as using some of these medications during the study will increase the risk of bleeding and also, may confuse pain evaluation as it is difficult to know how much pain relief was due to the study medication.
• To manage your knee or other pain you are allowed to use Panadol or Panadol Osteo with a total daily dose to not exceed 3000 mg per day. You are allowed to take Panadol to treat worsening pain, whether or not that pain is associated with OA of the index knee (e.g., may be used for example to treat a headache, acute injury, etc.).
• You will not be able to participate if you are taking statins prescribed to control your cholesterol level
The study coordinator will discuss these with you in detail.
Knee X-ray
We will be reviewing your knee x-ray for the presence of radiographic features associated with OA. It is sufficient for us if you can provide a recent knee x-ray image. It should be performed within the past 12 months from screening. Alternatively, you will be referred to Castlereagh Imaging at St Leonards. You can attend at your convenience within the first two weeks of the screening period.
Blood Test
As part of the assessment of your eligibility it will be necessary to perform a fasting blood test to check your overall health and cholesterol level. The blood test can be done at any of Pathology North Collection Centres within the Northern Sydney Local Health District. One of the centres is located at Royal North Shore Hospital.
If you are referred for a knee x-ray and blood test, both can be done on the same occasion as both centres are in St Leonards, and within 5 min walk from each other. You do not need an appointment for an x-ray or blood test.
You will be eligible for the study if you meet all the Inclusion criteria. You will not be eligible for the study if you meet any of the Exclusion criteria.
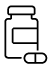
2. How much of my time will the study take?
Please refer to the following study visits diagram for more details regarding the time spent at each study event.

Index Knee MRI examination and Ultrasound scan test
Before starting the treatment (baseline visit), you will be scheduled for MRI scan (Castlereagh Imaging at Cremorne). This is to check for the presence of synovitis (knee inflammation) and bone marrow lesion/s (bone oedema). If both of your knees have OA, MRI scan will be performed on the most symptomatic knee identified at screening.
Ultrasound of your study knee will be performed after completion of your baseline assessments. It will be performed at Castlereagh Imaging Centre, St Leonards within 5 min walking from RNSH.
You will be required to complete a few surveys during the study duration.
Study Surveys
You will have approximately 35 mL (approximately 2.5 tablespoons) of blood taken at any given visit. The total amount of blood drawn during the whole study will be about 150 mL (approximately 10 tablespoons). All study visits require you in the fasting state.
You will be asked to provide a urine sample, which will be used to check your general health and pregnancy test (for female participants of childbearing potential only).
The faecal occult blood test (FOBT) is a lab test used to check stool samples for hidden (occult) blood. As part of the safety, you will be asked to provide small stool samples to monitor for risk of bleeding.
Blood, Urine and FOB Tests
You will be receiving a 5-week supply of PPS / placebo capsules as per your group allocation, to be taken at the recommended oral dose once every 4 days. Your study dose / capsules will be calculated by the study investigator and will be based on your body weight at baseline. Eating and drinking (e.g. milk, tea, coffee) interfere with the absorption of the study medication and so it is very important that you do not eat or drink for one hour before taking the medication and that you take study medication at least two hours after meals.
Study Treatment
PART 3 How will I manage my concerns?
1. ‘How will I manage my recommended oral dose’?
You will be sent an automatic notifications by SMS to manage your oral dose and remind you to take your study medication. It will help you to maintain a dosing schedule of the study medicine on every 4th day. Your dosage frequency would be pre-calculated from the baseline visit until the end of the study. You will be required to confirm use of study medication by completing weekly short surveys.
2. ‘How is this study funded to conduct the trial?’
The study is being sponsored by the University of Sydney and funded by a pharmaceutical company, Arthropharm Pty Ltd. The researchers involved in the study are not employees of Arthropharm and will not benefit financially from any of the findings of the study. There is no conflict of interest to be disclosed.
All the funds being paid by the pharmaceutical company to run the trial will be deposited into an account managed by the University of Sydney. No money is paid directly to individual researchers.
3. ‘What are the alternatives to participating in this study?’
If you decide not to participate in this study, and you wish to continue treatment, you will still receive the standard treatment available for your condition. It is important that you discuss the alternatives to participating in this study with your doctors.
4. ‘Are there risks to me in taking part in this study?’
All medical procedures involve some risk of injury. Also, there may be risks associated with this study that are presently unknown or unforeseeable. Possible risks are outlined below.
Pentosan Polysulfate (ELMIRON®)
Elmiron is contra-indicated (warned against) in patients with:
- A known hypersensitivity to PPS or related compounds
- Haemophilia
- An active (or recent history of) bleeding, alcoholic patients
- Concurrent heparin or oral anti-coagulant therapy
Warnings
Pregnancy – Reproduction studies performed in mice, rats and rabbits revealed no evidence of any potential for impaired fertility or harm to the foetus. An overall evaluation of these studies indicated minimal risk to the human foetus by the oral ingestion of up to 600mg Elmiron per day.
Data from studies in pregnant women undergoing abortion have indicated that PPS does not pass the placental barrier. There are, however, no well-controlled studies in pregnant women. Because animal studies are not always predictive of human response, Elmiron should not be used during pregnancy.
Nursing Mothers – It is not known if Elmiron is secreted in milk of animals or humans. Because many drugs are known to be excreted in human milk, Elmiron should not be administered to nursing women.
Paediatric Use – Safety and efficacy in children (<18 years) has not been established.
Other considerations
Habit-forming potential – Nil. Elmiron is not addictive.
Overdosage – no known reported incidents.
Very long-term continuous daily dosing (greater than 7 years, over 1.5 kg PPS accumulated dose) has been linked with a degenerative maculopathy in 20% of patients with interstitial cystitis, however the causative mechanism is yet to be established.
Some people may experience mild effects of swelling, headache, dizziness, nausea, indigestion, or diarrhoea. If any of these occur and are bothersome, contact the research coordinator on 02 9463 1896. Other unwanted effects not listed may also occur in some patients. Tell your doctor if you notice any other effects.
Precautions
We recommend avoiding high impact sports activities that can easily result in injury. Note that you can still exercise for example walking, running, and swimming.
We recommend reducing consumption of alcohol to a minimum. Alcohol consumption above recommended levels increases the risk for major bleeding from an injury.
Minor bleeds like dental floss, electric razor, trimming your fingernails and toenails can be avoided. Seek medical help if you injure yourself.
Due to the nature of the medication uptake in our study (Study Flowchart), you will always be re-assessed after going through a 5-week medication cycle. Those visits will allow us to monitor for safety. Any side-effect will be thoroughly reviewed by the study investigator.
Blood Draws
Drawing blood from a vein may cause local pain, bruising, occasional light-headedness, fainting, and very rarely, infection at the site of the blood draw. The blood pressure cuff may cause discomfort or bruising to the upper arm. Stopping the bleeding may take slightly longer than usual.
Radiation
As part of everyday living, everyone is exposed to naturally occurring background radiation and receives a dose of about 2 millisieverts (mSv) each year. The additional effective dose participants will receive from entering this trial is approximately 0.04 mSv. At this dose level, no harmful effects of radiation have been demonstrated as any effect is too small to measure. Studies suggest any risk is minimal.
Magnetic resonance imaging (MRI)
MRI stands for magnetic resonance imaging. An MRI scanner is a machine that uses electromagnetic radiation (radio waves) in a strong magnetic field to take clear pictures of the inside of the body. Electromagnetic radiation is not the same as ionising radiation used, for example, in X-rays. The images taken by the machine are called MRI scans.
There are no proven long-term risks related to MRI scans as used in this research project. MRI is considered to be safe when performed at a centre with appropriate procedures. However, the magnetic attraction for some metal objects can pose a safety risk, so it is essential to tell your us if you have:
− implanted pacemaker or defibrillator ;
− presence of metallic bodies in the eye;
− implantable hearing aids;
− insulin pumps;
− claustrophobia (a fear of being in closed or small spaces);
5. ‘Will participating in this study affect my plans to start a family?’
The effect of the study drug on an unborn baby is unknown. It is possible that the treatment regime under this study could have serious effects on the development of a foetus.
If at any time you think that either you or your sexual partner may be pregnant, it is vital to let researchers, or your medical team know immediately.
It is crucial that you are not pregnant and do not become pregnant during this study as the study drugs and procedures may damage an unborn baby.
If you are of childbearing potential and there is any possibility that you are pregnant, the researchers will need to perform a urine pregnancy test before you start in the study. If necessary, you must use reliable contraception (such as oral or implanted contraception, an IUD or have had a tubal ligation, or use condoms) during the course of this study.
6. ‘What happens if I suffer injury or complications as a result of the study?’
If you suffer any injuries or complications as a result of this study, you should contact the study investigator as soon as possible, who will assist you in arranging appropriate medical treatment.
You are also advised to take the national emergency services by dialling “000” for any untoward medical incident which requires immediate attention.
You will be provided with a Participant Identification Card stating that you are taking part in a clinical trial, the contact details of the study coordinator, and what to do in emergencies. You will always be required to keep this card with you during the study duration so that any physician can note your involvement with the study. This may be useful in the case of any complications that may arise, related or unrelated to the study.
You may have a right to take legal action to obtain compensation for any injuries or complications resulting from the study. Compensation may be available if your injury or complication is caused by the drugs or procedures, or by the negligence of any of the parties involved in the study. If you receive compensation that includes an amount for medical expenses, you will be required to pay for your medical treatment from those compensation monies.
If you are not eligible for compensation for your injury or complication under the law but are eligible for Medicare, then you can receive any medical treatment required for your injury or complication free of charge as a public patient in any Australian public hospital.
7. ‘Will I benefit from the study?’
This study aims to further medical knowledge and may improve future treatment of knee osteoarthritis; however, it may not directly benefit you. You may or may not experience an improvement in your OA symptoms by the end of the study. We cannot guarantee or promise that you will receive any benefits from this research; however, possible benefits may include reduced knee pain and improved knee function.
8. ‘Will taking part in this study cost me anything, and will I be paid?
Aside from giving your time, we do not expect that there will be any major costs associated with taking part in this study. Visit to the study site by public transport will not be compensated. We will cover the cost of all study-related procedures and examinations.
We will also provide you meal vouchers on every visit (for Zouki Café within RNSH) and a free parking voucher for all the study visits for your parking expenses at Parking P1 and P2 within hospital. Castlereagh Imaging centres at St. Leonards and Cremorne have their independent car parking facility. You can use the parking at these centres at no further expense.
Remote travellers may be reimbursed for travel expenses such as petrol costs with a grocery/gift vouchers.
9. ‘What will happen to my tissue samples after it has been used?’
The blood and urine samples provided during the study will be tested at the NSW Health Pathology laboratory to monitor the safety of study participants. A set of blood and urine samples will be collected for the bone and collagen turnover markers testing. Some of the tests will be performed by Arthropharm Pty Ltd, and some serum samples will be sent to Duke University (USA) for proteomic biomarkers testing that have shown to predict osteoarthritis progression better than traditional research methods.
Testing for bone and collagen turnover markers or proteomic biomarkers are not routinely performed by the NSLHD pathology labs, but all samples sent for testing will be de-identified (coded by Study ID and DOB) so your personal information will remain confidential.
The remaining set of samples will be stored at -80 degrees C at Royal North Shore Hospital as a backup to address the risk of specimen transportation. For any future research exploratory biomarkers not disclosed here, an ethics amendment would be sought for future testing. If the unused samples are not utilized within 2 years of the period after study completion, all the samples will be destroyed.
10. ‘How will my confidentiality be protected?’
By signing the consent form, you consent to the Study Investigator and relevant research staff collecting and using personal information about you for the research project. Any information obtained in connection with this research project that can identify you will remain confidential and will only be used for this research project. It will only be disclosed with your permission, except as required by law.
To ensure that your personal information is kept confidential, your name and any other information that allows you to be identified directly will not be entered on any records or samples the study coordinator provides to the Sponsor or the Sponsor’s authorised representatives. Instead, you will only be identified by a code (Study ID). The code is used so that you can be re-identified if necessary. The information collected about you will be held on password protected database (REDCap).
REDCap is a secure, web-based application designed to support data capture for research studies. It is hosted at the University of Sydney servers. The hard copies of your blood test reports will be kept in locked filing cabinets within Rheumatology Department, Royal North Shore Hospital accessible by the designated study coordinators.
The Sponsor or its authorised representatives will perform some of the tests, but all samples will only be identifiable by Study ID and DOB.
Any information obtained for this research project that can identify you will be treated as confidential and securely stored in accordance with Australian privacy laws for a minimum of 15 years after the completion of the research project. It will be disclosed only with your permission, or as required by law.
Information about you may be obtained from your health records held at this and other health services for the purpose of this research. By signing the consent form, you agree to the study team accessing health records if they are relevant to your participation in this research project. Information about your participation in this research project may also be recorded in your health records.
Your health records and any information obtained during the study may be inspected (for the purposes of verifying the procedures and the data) by the Food and Drug Administration (FDA) or by the Australian Government’s Therapeutic Goods Administration (TGA) or/and by the Human Research Ethics Committee (HREC) approving this study.
By signing this consent form, you authorise the release of, or access to, this confidential information to the relevant study personnel and regulatory authorities as noted above.
In accordance with relevant Australian and/or NSW privacy and other applicable laws, you always have the right to access and rectify your data or obtain a copy of your data collected. If your personal details are inaccurate or incomplete or are not being processed in compliance with the applicable regulatory requirements, you may ask for a correction or to block your personal data. Please contact the study team member named at the end of this document if you would like to access your information. If you participate in this trial, you will not own any of the information collected or produced for the purpose of the trial.
A description of this clinical trial is available on Australian New Zealand Clinical Trial Registry https://www.anzctr.org.au (#: ACTRN12621000654853)
This web site will not include information that can identify you. At most, the web site will include a summary of the results. You can search this web site at any time.
11. ‘What happens with the results?’
If you give us your permission by signing the consent document, we plan to discuss/publish the results in journals, presentations at conferences, symposia, Congress and scientific meetings in general. No information which could lead to your identification will be included in the dissemination of results.
In any publication, information will be provided in such a way that you cannot be identified.
13. Can I tell other people about the study?
Yes, you are welcome to tell other people about the study by sharing the link sent to you.
14. ‘What happened to my treatment when the study is finished?’
You will not be supplied the study drug following completion of this study. Any decision to take PPS after the trial should be made in consultation with you and your treating doctor considering the most appropriate treatment for you at that time.
15. ‘‘What should I do if I want to discuss this study further before I decide?’
When you have read this information, the researcher coordinator will discuss it with you and any queries you may have. If you would like to know more at any stage, please do not hesitate to contact the study coordinator on 02 9463 1896.
16. ‘Who should I contact if I have concerns about the conduct of this study?’
This study has been approved by the Northern Sydney Local Health District HREC. Any person with concerns or complaints about the conduct of this study should contact the Research Office who is nominated to receive complaints from research participants.
You should contact the following:
Local Complaints Contact Person
Name
Research Office
Position
Research Governance Officer
Telephone
02 9926 4590
NSLHD-Research@health.nsw.gov.au
Reference
2021/ETH00315
Thank you for taking the time to consider this study.
To download a copy of the Participant Information Sheet click here.